2_Goseq analysis & IRE enrichment test
Yang & Steve
24/02/2020
Last updated: 2020-03-17
Checks: 7 0
Knit directory: 20190717_Lardelli_RNASeq_Larvae/
This reproducible R Markdown analysis was created with workflowr (version 1.6.0). The Checks tab describes the reproducibility checks that were applied when the results were created. The Past versions tab lists the development history.
Great! Since the R Markdown file has been committed to the Git repository, you know the exact version of the code that produced these results.
Great job! The global environment was empty. Objects defined in the global environment can affect the analysis in your R Markdown file in unknown ways. For reproduciblity it’s best to always run the code in an empty environment.
The command set.seed(20200227) was run prior to running the code in the R Markdown file. Setting a seed ensures that any results that rely on randomness, e.g. subsampling or permutations, are reproducible.
Great job! Recording the operating system, R version, and package versions is critical for reproducibility.
Nice! There were no cached chunks for this analysis, so you can be confident that you successfully produced the results during this run.
Great job! Using relative paths to the files within your workflowr project makes it easier to run your code on other machines.
Great! You are using Git for version control. Tracking code development and connecting the code version to the results is critical for reproducibility. The version displayed above was the version of the Git repository at the time these results were generated.
Note that you need to be careful to ensure that all relevant files for the analysis have been committed to Git prior to generating the results (you can use wflow_publish or wflow_git_commit). workflowr only checks the R Markdown file, but you know if there are other scripts or data files that it depends on. Below is the status of the Git repository when the results were generated:
Ignored files:
Ignored: .DS_Store
Ignored: .Rhistory
Ignored: .Rproj.user/
Ignored: 1_DE-gene-analysis_cache/
Ignored: 1_DE-gene-analysis_files/
Ignored: analysis/.DS_Store
Ignored: analysis/.Rhistory
Ignored: analysis/.Rproj.user/
Ignored: data/.DS_Store
Ignored: data/0_rawData/.DS_Store
Ignored: data/1_trimmedData/.DS_Store
Ignored: data/2_alignedData/.DS_Store
Ignored: files/
Ignored: output/.DS_Store
Note that any generated files, e.g. HTML, png, CSS, etc., are not included in this status report because it is ok for generated content to have uncommitted changes.
These are the previous versions of the R Markdown and HTML files. If you’ve configured a remote Git repository (see ?wflow_git_remote), click on the hyperlinks in the table below to view them.
| File | Version | Author | Date | Message |
|---|---|---|---|---|
| Rmd | 13abd74 | yangdongau | 2020-03-17 | Use logFC replace rank statistic in pathview. |
| Rmd | fa3b603 | yangdongau | 2020-03-04 | 1.Reorganize the files |
| html | fa3b603 | yangdongau | 2020-03-04 | 1.Reorganize the files |
| Rmd | 2556481 | yangdongau | 2020-03-04 | Add description |
| Rmd | 3c8b6e1 | yangdongau | 2020-02-28 | Add in annotations. |
| Rmd | 8f91594 | yangdongau | 2020-02-28 | Add in annotations. |
| Rmd | cfc2be0 | yangdongau | 2020-02-27 | clean up library |
| Rmd | dc5cbe9 | yangdongau | 2020-02-27 | rename&clean up packages |
Setup
library(limma)
library(edgeR)
library(AnnotationHub)
library(tidyverse)
library(magrittr)
library(pander)
library(ggrepel)
library(scales)
library(org.Hs.eg.db)
library(plyr)
library(ggraph)
library(tidygraph)
library(fgsea)
library(goseq)
library(org.Dr.eg.db)
library(msigdbr)
library(rWikiPathways)
theme_set(theme_bw())
panderOptions("big.mark", ",")
panderOptions("table.split.table", Inf)
panderOptions("table.style", "rmarkdown")
if (interactive()) setwd(here::here("analysis"))Annotation was set up as a EnsDb object based on Ensembl Release 96.
ah <- AnnotationHub() %>%
subset(species == "Danio rerio") %>%
subset(dataprovider == "Ensembl") %>%
subset(rdataclass == "EnsDb")
ensDb <- ah[["AH69169"]]
genesGR <- genes(ensDb)
transGR <- transcripts(ensDb)
DrEns2Symbol <- genesGR %>%
mcols() %>%
as_tibble() %>%
dplyr::select(gene_id, gene_name)Data load
dgeList <- read_rds(here::here("data","dgeList.rds"))
entrezGenes <- dgeList$genes %>%
dplyr::filter(!is.na(entrez_gene)) %>%
unnest(entrez_gene) %>%
dplyr::rename(entrez_gene = entrez_gene)
topTable <- file.path(here::here("output", "topTable.csv")) %>%
read_csv()
topTableDE <- file.path(here::here("output", "DEgenes.csv")) %>%
read_csv()Databases used for testing
Hallmark and KEGG pathway gene mappings were achieved by msigdbr, and Wiki pathway gene mapping was downloaded by rWikiPathways.
hallmark <- msigdbr("Danio rerio", category = "H") %>%
left_join(entrezGenes) %>%
dplyr::filter(!is.na(ensembl_gene_id)) %>%
distinct(gs_name, ensembl_gene_id, .keep_all = TRUE)
hallmarkByGene <- hallmark %>%
split(f = .$ensembl_gene_id) %>%
lapply(extract2, "gs_name")
hallmarkByID <- hallmark %>%
split(f = .$gs_name) %>%
lapply(extract2, "ensembl_gene_id")kegg <- msigdbr("Danio rerio", category = "C2", subcategory = "CP:KEGG") %>%
left_join(entrezGenes) %>%
dplyr::filter(!is.na(ensembl_gene_id)) %>%
distinct(gs_name, ensembl_gene_id, .keep_all = TRUE)
keggByGene <- kegg %>%
split(f = .$ensembl_gene_id) %>%
lapply(extract2, "gs_name")
keggByID <- kegg %>%
split(f = .$gs_name) %>%
lapply(extract2, "ensembl_gene_id")wikidownload <- downloadPathwayArchive(organism = "Danio rerio", format = "gmt")
wiki <- gmtPathways(here::here("analysis","wikipathways-20200210-gmt-Danio_rerio.gmt"))
wikilist <- names(wiki) %>%
lapply(function(x){
tibble(pathway = x, entrez_gene = wiki[[x]])
}) %>%
bind_rows() %>%
mutate(entrez_gene = as.numeric(entrez_gene)) %>%
left_join(entrezGenes) %>%
dplyr::filter(!is.na(ensembl_gene_id)) %>%
distinct(pathway, ensembl_gene_id, .keep_all = TRUE) %>%
mutate(pathway = str_remove_all(pathway, "%.+"))
wikiByGene <- wikilist %>%
split(f = .$ensembl_gene_id) %>%
lapply(extract2, "pathway")
wikiByID <- wikilist %>%
split(f = .$pathway) %>%
lapply(extract2, "ensembl_gene_id")GOseq analysis
Calculate pwf using gene lengths as bia data.
pwf <- topTable %>%
dplyr::select(ensembl_gene_id, logFC, DE, aveLength, aveGc) %>%
mutate(nGC = aveLength*aveGc) %>%
distinct(ensembl_gene_id, .keep_all = TRUE) %>%
with(
nullp(
DEgenes = structure(
as.integer(DE), names = ensembl_gene_id
),
id = "ensGene",
# bias.data = log(nGC),
bias.data = aveLength,
plot.fit = FALSE
)
)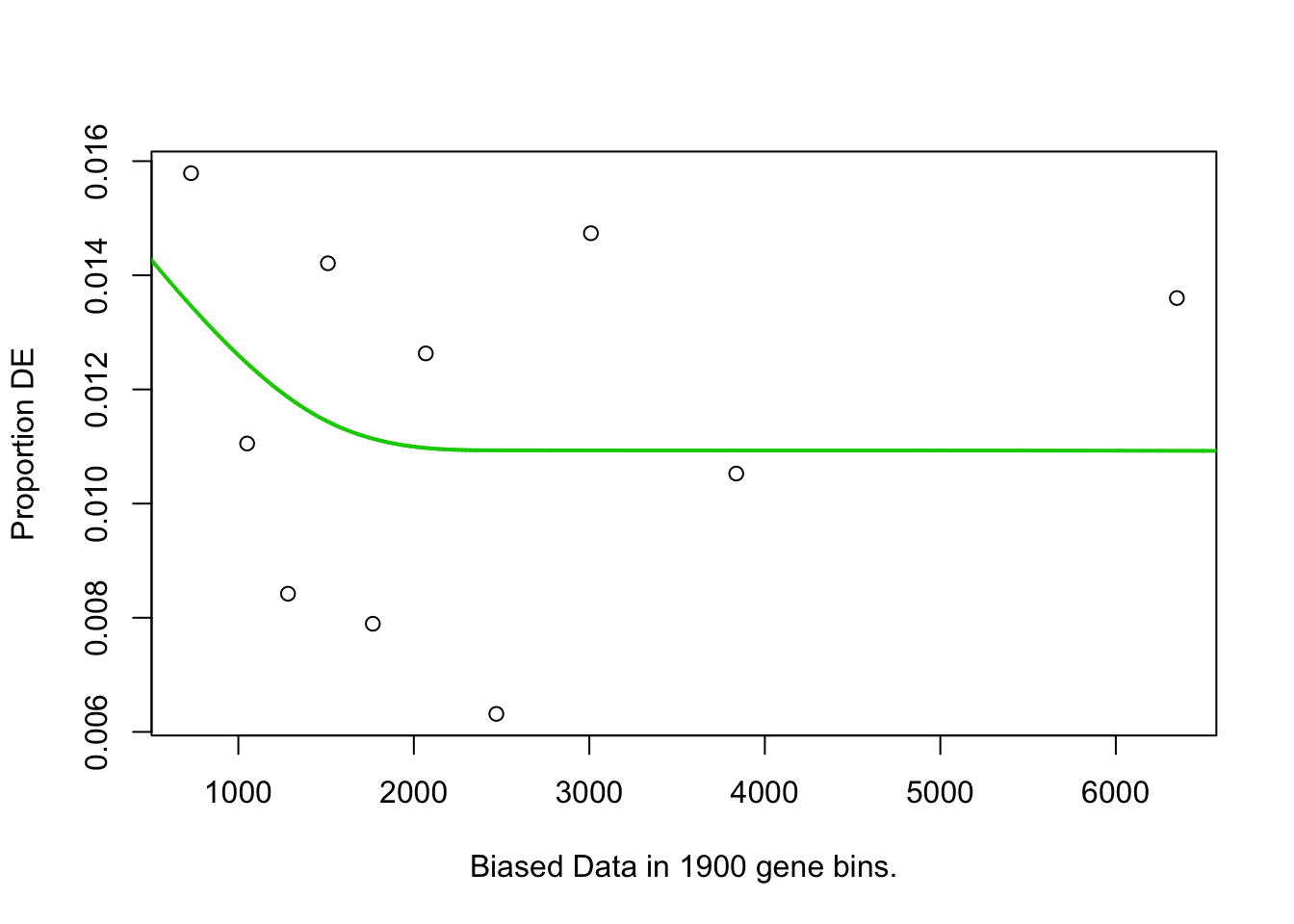
Probability weight function for a gene being considered as DE based on the number of GC bases per gene.
| Version | Author | Date |
|---|---|---|
| fa3b603 | yangdongau | 2020-03-04 |
Goseq pathway analysis
Use goseq to perform pathway enrichment analysis and print out the top 5 significantly-changed pathways
Hallmark Gene Sets
hallmarkGoseq <- pwf %>%
goseq(gene2cat = hallmarkByGene) %>%
as_tibble %>%
mutate(FDR = p.adjust(over_represented_pvalue, method = "fdr"))
hallmarkGoseq %>%
dplyr::slice(1:5) %>%
dplyr::select(
Pathway = category,
starts_with("num"),
PValue = over_represented_pvalue,
FDR
) %>%
pander(
caption = "Most highly ranked hallmark pathways.",
justify = "lrrrr"
)| Pathway | numDEInCat | numInCat | PValue | FDR |
|---|---|---|---|---|
| HALLMARK_G2M_CHECKPOINT | 19 | 182 | 2.736e-12 | 1.368e-10 |
| HALLMARK_E2F_TARGETS | 13 | 174 | 1.011e-06 | 2.528e-05 |
| HALLMARK_MYC_TARGETS_V1 | 8 | 197 | 0.009651 | 0.1609 |
| HALLMARK_ESTROGEN_RESPONSE_EARLY | 6 | 194 | 0.06376 | 0.5024 |
| HALLMARK_MITOTIC_SPINDLE | 6 | 199 | 0.06515 | 0.5024 |
KEGG Pathways
keggGoseq <- pwf %>%
goseq(gene2cat = keggByGene) %>%
as_tibble %>%
mutate(FDR = p.adjust(over_represented_pvalue, method = "fdr"))
keggGoseq %>%
dplyr::slice(1:5) %>%
dplyr::select(
Pathway = category,
starts_with("num"),
PValue = over_represented_pvalue,
FDR
) %>%
pander(
caption = "Most highly ranked KEGG pathways.",
justify = "lrrrr"
)| Pathway | numDEInCat | numInCat | PValue | FDR |
|---|---|---|---|---|
| KEGG_DNA_REPLICATION | 7 | 34 | 2.241e-08 | 4.169e-06 |
| KEGG_CELL_CYCLE | 8 | 109 | 5.691e-06 | 0.0005293 |
| KEGG_FATTY_ACID_METABOLISM | 3 | 43 | 0.007278 | 0.3496 |
| KEGG_PRIMARY_BILE_ACID_BIOSYNTHESIS | 2 | 16 | 0.009276 | 0.3496 |
| KEGG_ARGININE_AND_PROLINE_METABOLISM | 3 | 47 | 0.009399 | 0.3496 |
wiki pathway
wikiGoseq <- pwf %>%
goseq(gene2cat = wikiByGene) %>%
as_tibble %>%
mutate(FDR = p.adjust(over_represented_pvalue, method = "fdr"))
wikiGoseq %>%
dplyr::slice(1:5) %>%
dplyr::select(
Pathway = category,
starts_with("num"),
PValue = over_represented_pvalue,
FDR
) %>%
pander(
caption = "Most highly ranked wiki pathways.",
justify = "lrrrr"
)| Pathway | numDEInCat | numInCat | PValue | FDR |
|---|---|---|---|---|
| DNA Replication | 6 | 31 | 5.138e-07 | 4.265e-05 |
| Cell cycle | 7 | 71 | 5.006e-06 | 0.0002078 |
| G1 to S cell cycle control | 6 | 49 | 8.095e-06 | 0.000224 |
| Effect of L-carnitine on metabolism | 2 | 23 | 0.02278 | 0.4728 |
| Endochondral Ossification | 2 | 41 | 0.06718 | 1 |
Goseq GO analysis
goByGene <- links(org.Dr.egGO2ALLEGS) %>%
as_tibble() %>%
left_join(
links(org.Dr.egENSEMBL2EG)
) %>%
distinct(ensembl_id, go_id) %>%
dplyr::filter(ensembl_id %in% topTable$ensembl_gene_id) %>%
split(f = .$ensembl_id) %>%
lapply(extract2, "go_id")
goGoseq <- pwf %>%
goseq(gene2cat = goByGene) %>%
as_tibble %>%
mutate(FDR = p.adjust(over_represented_pvalue, method = "fdr"))Enriched GO list is filter by a FDR cutoff of 0.05.
goGoseq %>%
dplyr::filter(FDR < 0.05) %>%
dplyr::select(
Pathway = category,
starts_with("num"),
term,
ontology,
PValue = over_represented_pvalue,
FDR
) %>%
pander(
caption = "Most highly ranked GO pathways.",
justify = "lrrrrrr"
)| Pathway | numDEInCat | numInCat | term | ontology | PValue | FDR |
|---|---|---|---|---|---|---|
| GO:0042555 | 7 | 9 | MCM complex | CC | 5.973e-13 | 7.943e-09 |
| GO:0006267 | 6 | 7 | pre-replicative complex assembly involved in nuclear cell cycle DNA replication | BP | 1.264e-11 | 4.204e-08 |
| GO:0036388 | 6 | 7 | pre-replicative complex assembly | BP | 1.264e-11 | 4.204e-08 |
| GO:1902299 | 6 | 7 | pre-replicative complex assembly involved in cell cycle DNA replication | BP | 1.264e-11 | 4.204e-08 |
| GO:0071103 | 12 | 74 | DNA conformation change | BP | 2.789e-11 | 7.42e-08 |
| GO:0000727 | 6 | 9 | double-strand break repair via break-induced replication | BP | 1.394e-10 | 3.089e-07 |
| GO:0007049 | 27 | 598 | cell cycle | BP | 6.409e-10 | 1.218e-06 |
| GO:0006260 | 12 | 100 | DNA replication | BP | 1.268e-09 | 2.108e-06 |
| GO:0033260 | 6 | 13 | nuclear DNA replication | BP | 2.927e-09 | 4.326e-06 |
| GO:0003688 | 6 | 14 | DNA replication origin binding | MF | 4.667e-09 | 6.206e-06 |
| GO:0000278 | 18 | 309 | mitotic cell cycle | BP | 1.117e-08 | 1.351e-05 |
| GO:0044786 | 6 | 16 | cell cycle DNA replication | BP | 1.316e-08 | 1.433e-05 |
| GO:0005198 | 23 | 488 | structural molecule activity | MF | 1.401e-08 | 1.433e-05 |
| GO:0006270 | 6 | 17 | DNA replication initiation | BP | 1.874e-08 | 1.781e-05 |
| GO:0032508 | 6 | 17 | DNA duplex unwinding | BP | 2.042e-08 | 1.81e-05 |
| GO:0032392 | 6 | 18 | DNA geometric change | BP | 2.993e-08 | 2.488e-05 |
| GO:0022402 | 19 | 374 | cell cycle process | BP | 3.744e-08 | 2.78e-05 |
| GO:0006261 | 9 | 64 | DNA-dependent DNA replication | BP | 3.763e-08 | 2.78e-05 |
| GO:0017116 | 4 | 5 | single-stranded DNA-dependent ATP-dependent DNA helicase activity | MF | 7.361e-08 | 5.153e-05 |
| GO:0008094 | 8 | 57 | DNA-dependent ATPase activity | MF | 2.013e-07 | 0.0001339 |
| GO:0006310 | 10 | 101 | DNA recombination | BP | 2.139e-07 | 0.0001355 |
| GO:0006259 | 18 | 374 | DNA metabolic process | BP | 2.318e-07 | 0.0001402 |
| GO:0071824 | 9 | 83 | protein-DNA complex subunit organization | BP | 3.804e-07 | 0.00022 |
| GO:0065004 | 8 | 62 | protein-DNA complex assembly | BP | 4.571e-07 | 0.0002533 |
| GO:0043142 | 4 | 8 | single-stranded DNA-dependent ATPase activity | MF | 1.002e-06 | 0.0005332 |
| GO:0140097 | 10 | 123 | catalytic activity, acting on DNA | MF | 1.166e-06 | 0.0005962 |
| GO:0003678 | 6 | 34 | DNA helicase activity | MF | 1.609e-06 | 0.0007926 |
| GO:0006268 | 4 | 9 | DNA unwinding involved in DNA replication | BP | 1.684e-06 | 0.0007973 |
| GO:1902969 | 4 | 9 | mitotic DNA replication | BP | 1.738e-06 | 0.0007973 |
| GO:0006281 | 13 | 232 | DNA repair | BP | 2.145e-06 | 0.000951 |
| GO:0000724 | 7 | 54 | double-strand break repair via homologous recombination | BP | 2.316e-06 | 0.0009625 |
| GO:0000725 | 7 | 54 | recombinational repair | BP | 2.316e-06 | 0.0009625 |
| GO:0005201 | 7 | 60 | extracellular matrix structural constituent | MF | 3.622e-06 | 0.00146 |
| GO:0006974 | 15 | 324 | cellular response to DNA damage stimulus | BP | 3.745e-06 | 0.001465 |
| GO:1902292 | 3 | 4 | cell cycle DNA replication initiation | BP | 4.841e-06 | 0.001707 |
| GO:1902315 | 3 | 4 | nuclear cell cycle DNA replication initiation | BP | 4.841e-06 | 0.001707 |
| GO:1902975 | 3 | 4 | mitotic DNA replication initiation | BP | 4.841e-06 | 0.001707 |
| GO:0051276 | 18 | 466 | chromosome organization | BP | 4.878e-06 | 0.001707 |
| GO:1903047 | 12 | 224 | mitotic cell cycle process | BP | 7.989e-06 | 0.002724 |
| GO:0004386 | 8 | 96 | helicase activity | MF | 1.039e-05 | 0.003453 |
| GO:0006302 | 8 | 95 | double-strand break repair | BP | 1.127e-05 | 0.003657 |
| GO:0004003 | 5 | 28 | ATP-dependent DNA helicase activity | MF | 1.159e-05 | 0.00367 |
| GO:0006271 | 4 | 14 | DNA strand elongation involved in DNA replication | BP | 1.485e-05 | 0.004589 |
| GO:0003697 | 6 | 47 | single-stranded DNA binding | MF | 1.518e-05 | 0.004589 |
| GO:0006323 | 6 | 50 | DNA packaging | BP | 1.794e-05 | 0.005301 |
| GO:0022616 | 4 | 15 | DNA strand elongation | BP | 1.978e-05 | 0.005633 |
| GO:0005212 | 6 | 45 | structural constituent of eye lens | MF | 1.991e-05 | 0.005633 |
| GO:0043138 | 4 | 17 | 3’-5’ DNA helicase activity | MF | 2.978e-05 | 0.008086 |
| GO:0099080 | 15 | 389 | supramolecular complex | CC | 3.04e-05 | 0.008086 |
| GO:0099081 | 15 | 389 | supramolecular polymer | CC | 3.04e-05 | 0.008086 |
| GO:0098813 | 7 | 88 | nuclear chromosome segregation | BP | 5.442e-05 | 0.01419 |
| GO:0006826 | 4 | 21 | iron ion transport | BP | 8.407e-05 | 0.0215 |
| GO:0099512 | 14 | 388 | supramolecular fiber | CC | 0.0001142 | 0.02865 |
| GO:1990518 | 2 | 2 | single-stranded DNA-dependent ATP-dependent 3’-5’ DNA helicase activity | MF | 0.0001363 | 0.03339 |
| GO:0033554 | 19 | 652 | cellular response to stress | BP | 0.0001381 | 0.03339 |
| GO:0007059 | 7 | 106 | chromosome segregation | BP | 0.0001794 | 0.0426 |
| GO:0000819 | 6 | 76 | sister chromatid segregation | BP | 0.0001992 | 0.04648 |
goGoseqTop <- goGoseq %>%
dplyr::filter(FDR < 0.05) %>%
dplyr::select(
Pathway = category,
starts_with("num"),
term,
ontology,
PValue = over_represented_pvalue,
FDR)GO plot
Use the links between DE genes and signficant GO terms to plot the GO networks.
## Get significant GO terms
sigGo <- goGoseq %>%
dplyr::filter(FDR < 0.05) %>%
.$category
## Convert list of GO terms by gene to list of genes by GO term
geneByGo <- names(goByGene) %>%
lapply(function(x){tibble(gene_id = x, go_id = goByGene[[x]])}) %>%
bind_rows() %>%
split(f = .$go_id) %>%
lapply(magrittr::extract2, "gene_id")
## Get DE genes that belong to sigificant GO terms
goGenes <- lapply(
sigGo,
function(x){
geneByGo[[x]][geneByGo[[x]] %in% topTableDE$ensembl_gene_id]
}
)
names(goGenes) <- sigGo
## Make tibble of GO terms
goTerms <- names(goGenes) %>%
tibble::enframe(name = NULL, value = "label")
## Make tibble of genes
genes <- unlist(goGenes) %>%
unique() %>%
tibble::enframe(name = NULL, value = "label") %>%
mutate
## Join to create node list
nodes <- rbind(goTerms, genes) %>%
rowid_to_column("id")
## Create edge list
edges <- goGenes %>%
stack() %>%
as_tibble() %>%
dplyr::select(goTerm = ind, geneId = values) %>%
dplyr::arrange(goTerm) %>%
mutate(goTerm = as.character(goTerm)) %>%
left_join(nodes, by = c("goTerm" = "label")) %>%
dplyr::rename(from = id) %>%
left_join(nodes, by = c("geneId" = "label")) %>%
dplyr::rename(to = id) %>%
dplyr::select(from, to)
## Setup colours
colours <- length(sigGo) %>%
rainbow()
## Create tidygraph object
tidy <- tbl_graph(
nodes = nodes,
edges = edges,
directed = FALSE
) %>%
activate(nodes) %>%
mutate(
goTerms = case_when(
id <= length(sigGo) ~ label
),
term = Term(label),
gene_id = case_when(
!label %in% sigGo ~ label
),
colour = case_when(
id <= length(sigGo) ~ colours[id]
),
size = ifelse(id <= length(sigGo), 4, 1)
) %>%
left_join(DrEns2Symbol) %>%
activate(edges) %>%
mutate(
colour = case_when(
from <= length(sigGo) ~ colours[from]
)
)
## Set seed to allow same graph to be produced each time function is executed
set.seed(1234)
## Plot network graph
ggraph(tidy, layout = "fr") +
scale_fill_manual(
values = "white",
na.value = "gray80"
) +
scale_edge_color_manual(
values = "black",
na.value = "gray80"
) +
geom_edge_arc(
aes(color = "black"),
alpha = 0.5,
show.legend = FALSE,
curvature = 0.5
) +
geom_node_point(
aes(fill = "black", size = size),
shape = 21,
stroke = 0.5,
show.legend = FALSE
) +
geom_node_label(
aes(label = gene_name),
repel = TRUE,
size = 3,
alpha = 0.7,
label.padding = 0.1
) +
theme_graph() +
theme(legend.position = "none")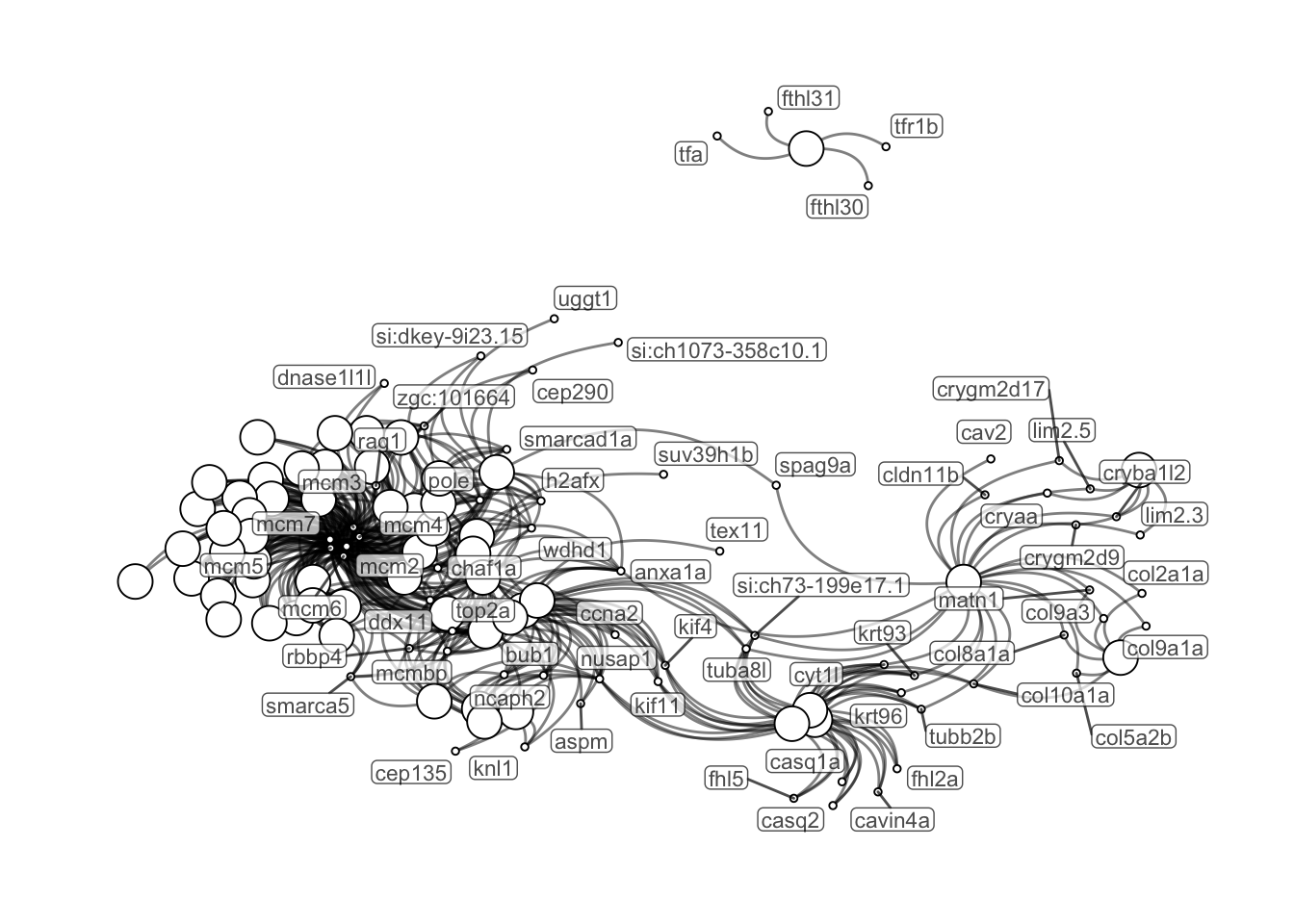
| Version | Author | Date |
|---|---|---|
| fa3b603 | yangdongau | 2020-03-04 |
IRE enrichment analysis
IRE Goseq analysis
ireGR <- list.files(
path = here::here("analysis"),
pattern = "gff.gz",
full.names = TRUE
) %>%
sapply(rtracklayer::import.gff, simplify = FALSE) %>%
lapply(function(x){
tibble(
tx_id = seqnames(x) %>% as.character(),
quality = x$quality
) %>%
left_join(
mcols(transGR) %>% as.data.frame()
) %>%
mutate(
quality = factor(
quality, levels = c("Low", "Medium", "High")
)
) %>%
arrange(gene_id, desc(quality)) %>%
distinct(gene_id, .keep_all = TRUE) %>%
dplyr::select(tx_id, gene_id, quality)
})
names(ireGR) <- str_extract(names(ireGR), "utr[35]")
ireHigh <- lapply(ireGR, subset, quality == "High")ireByGene <- c(
names(ireGR) %>%
lapply(function(x){
mutate(ireGR[[x]], Type = paste0(x, "_All"))
}),
names(ireHigh) %>%
lapply(function(x){
mutate(ireHigh[[x]], Type = paste0(x, "_High"))
})
) %>%
bind_rows() %>%
split(f = .$gene_id) %>%
lapply(function(x){
unique(x$Type)
})
ireGoseq <- pwf %>%
goseq(gene2cat = ireByGene) %>%
as_tibble %>%
mutate(FDR = p.adjust(over_represented_pvalue, method = "fdr"))| Pathway | numDEInCat | numInCat | PValue | FDR |
|---|---|---|---|---|
| utr5_All | 6 | 306 | 0.1824 | 0.7296 |
| utr5_High | 1 | 39 | 0.4081 | 0.8162 |
| utr3_High | 1 | 72 | 0.6138 | 0.8184 |
| utr3_All | 9 | 883 | 0.9472 | 0.9472 |
IRE GSEA analysis
Gene ranks
ranks <- topTable %>%
mutate(stat = -sign(logFC) * log10(PValue)) %>%
dplyr::arrange(stat) %>%
with(structure(stat, names = ensembl_gene_id))ireGSEA <- c(
names(ireGR) %>%
lapply(function(x){
mutate(ireGR[[x]], Type = paste0(x, "_All"))
}),
names(ireHigh) %>%
lapply(function(x){
mutate(ireHigh[[x]], Type = paste0(x, "_High"))
})
) %>%
bind_rows() %>%
split(f = .$Type) %>%
lapply(function(x){
unique(x$gene_id)
})
fgseaIRE <- fgsea(ireGSEA, ranks, nperm=1e5) %>%
as_tibble() %>%
dplyr::rename(FDR = padj) %>%
mutate(padj = p.adjust(pval, "bonferroni")) %>%
dplyr::arrange(pval)
fgseaIRE %>%
dplyr::select(-leadingEdge, -nMoreExtreme) %>%
pander(
style = "rmarkdown",
split.tables = Inf,
justify = "lrrrrrr",
caption = paste(
"The", nrow(.), "GSEA analysis of IRE enrichment", percent(max(.$FDR)))
)| pathway | pval | FDR | ES | NES | size | padj |
|---|---|---|---|---|---|---|
| utr3_High | 0.1514 | 0.5524 | -0.4077 | -1.231 | 72 | 0.6056 |
| utr5_High | 0.2762 | 0.5524 | 0.3656 | 1.115 | 39 | 1 |
| utr3_All | 0.6273 | 0.6663 | -0.2577 | -0.9628 | 883 | 1 |
| utr5_All | 0.6663 | 0.6663 | -0.2614 | -0.9256 | 306 | 1 |
Data export
Goseqpathway <- bind_rows(
hallmarkGoseq,
keggGoseq,
wikiGoseq
) %>%
dplyr::filter(FDR < 0.05) %>%
dplyr::select(
category, numDEInCat, numInCat, FDR
)
write_csv(Goseqpathway, here::here("output","Goseq_pathway.csv"))
# output IRE enrichment results
write_csv(ireGoseq,here::here("output","ireGoseq_resulst.csv"))
fgseaIREresult <- fgseaIRE %>%
dplyr::select(
pathway, ES, NES, size, padj
)
write_csv(fgseaIREresult,here::here("output","ireGSEA_resulst.csv"))
devtools::session_info()─ Session info ──────────────────────────────────────────────────────────
setting value
version R version 3.6.0 (2019-04-26)
os macOS Mojave 10.14.6
system x86_64, darwin15.6.0
ui X11
language (EN)
collate en_AU.UTF-8
ctype en_AU.UTF-8
tz Australia/Adelaide
date 2020-03-17
─ Packages ──────────────────────────────────────────────────────────────
package * version date lib source
AnnotationDbi * 1.46.1 2019-08-20 [1] Bioconductor
AnnotationFilter * 1.8.0 2019-05-02 [1] Bioconductor
AnnotationHub * 2.16.0 2019-05-02 [1] Bioconductor
assertthat 0.2.1 2019-03-21 [1] CRAN (R 3.6.0)
backports 1.1.4 2019-04-10 [1] CRAN (R 3.6.0)
BiasedUrn * 1.07 2015-12-28 [1] CRAN (R 3.6.0)
Biobase * 2.44.0 2019-05-02 [1] Bioconductor
BiocFileCache * 1.8.0 2019-05-02 [1] Bioconductor
BiocGenerics * 0.30.0 2019-05-02 [1] Bioconductor
BiocManager 1.30.4 2018-11-13 [1] CRAN (R 3.6.0)
BiocParallel 1.18.1 2019-08-06 [1] Bioconductor
biomaRt 2.40.4 2019-08-19 [1] Bioconductor
Biostrings 2.52.0 2019-05-02 [1] Bioconductor
bit 1.1-14 2018-05-29 [1] CRAN (R 3.6.0)
bit64 0.9-7 2017-05-08 [1] CRAN (R 3.6.0)
bitops 1.0-6 2013-08-17 [1] CRAN (R 3.6.0)
blob 1.2.0 2019-07-09 [1] CRAN (R 3.6.0)
broom 0.5.2 2019-04-07 [1] CRAN (R 3.6.0)
callr 3.3.1 2019-07-18 [1] CRAN (R 3.6.0)
caTools 1.17.1.2 2019-03-06 [1] CRAN (R 3.6.0)
cellranger 1.1.0 2016-07-27 [1] CRAN (R 3.6.0)
cli 1.1.0 2019-03-19 [1] CRAN (R 3.6.0)
colorspace 1.4-1 2019-03-18 [1] CRAN (R 3.6.0)
crayon 1.3.4 2017-09-16 [1] CRAN (R 3.6.0)
curl 4.0 2019-07-22 [1] CRAN (R 3.6.0)
data.table 1.12.2 2019-04-07 [1] CRAN (R 3.6.0)
DBI 1.0.0 2018-05-02 [1] CRAN (R 3.6.0)
dbplyr * 1.4.2 2019-06-17 [1] CRAN (R 3.6.0)
DelayedArray 0.10.0 2019-05-02 [1] Bioconductor
desc 1.2.0 2018-05-01 [1] CRAN (R 3.6.0)
devtools 2.2.2 2020-02-17 [1] CRAN (R 3.6.0)
digest 0.6.20 2019-07-04 [1] CRAN (R 3.6.0)
dplyr * 0.8.3 2019-07-04 [1] CRAN (R 3.6.0)
edgeR * 3.26.7 2019-08-13 [1] Bioconductor
ellipsis 0.3.0 2019-09-20 [1] CRAN (R 3.6.0)
ensembldb * 2.8.0 2019-05-02 [1] Bioconductor
evaluate 0.14 2019-05-28 [1] CRAN (R 3.6.0)
farver 1.1.0 2018-11-20 [1] CRAN (R 3.6.0)
fastmatch 1.1-0 2017-01-28 [1] CRAN (R 3.6.0)
fgsea * 1.10.1 2019-08-21 [1] Bioconductor
forcats * 0.4.0 2019-02-17 [1] CRAN (R 3.6.0)
fs 1.3.1 2019-05-06 [1] CRAN (R 3.6.0)
geneLenDataBase * 1.20.0 2019-05-07 [1] Bioconductor
generics 0.0.2 2018-11-29 [1] CRAN (R 3.6.0)
GenomeInfoDb * 1.20.0 2019-05-02 [1] Bioconductor
GenomeInfoDbData 1.2.1 2019-07-25 [1] Bioconductor
GenomicAlignments 1.20.1 2019-06-18 [1] Bioconductor
GenomicFeatures * 1.36.4 2019-07-09 [1] Bioconductor
GenomicRanges * 1.36.0 2019-05-02 [1] Bioconductor
ggforce 0.3.1 2019-08-20 [1] CRAN (R 3.6.0)
ggplot2 * 3.2.1 2019-08-10 [1] CRAN (R 3.6.0)
ggraph * 1.0.2 2018-07-07 [1] CRAN (R 3.6.0)
ggrepel * 0.8.1 2019-05-07 [1] CRAN (R 3.6.0)
git2r 0.26.1 2019-06-29 [1] CRAN (R 3.6.0)
glue 1.3.1 2019-03-12 [1] CRAN (R 3.6.0)
GO.db 3.8.2 2019-08-09 [1] Bioconductor
goseq * 1.36.0 2019-05-02 [1] Bioconductor
gridExtra 2.3 2017-09-09 [1] CRAN (R 3.6.0)
gtable 0.3.0 2019-03-25 [1] CRAN (R 3.6.0)
haven 2.1.1 2019-07-04 [1] CRAN (R 3.6.0)
here 0.1 2017-05-28 [1] CRAN (R 3.6.0)
highr 0.8 2019-03-20 [1] CRAN (R 3.6.0)
hms 0.5.1 2019-08-23 [1] CRAN (R 3.6.0)
htmltools 0.3.6 2017-04-28 [1] CRAN (R 3.6.0)
httpuv 1.5.1 2019-04-05 [1] CRAN (R 3.6.0)
httr 1.4.1 2019-08-05 [1] CRAN (R 3.6.0)
igraph 1.2.4.1 2019-04-22 [1] CRAN (R 3.6.0)
interactiveDisplayBase 1.22.0 2019-05-02 [1] Bioconductor
IRanges * 2.18.2 2019-08-24 [1] Bioconductor
jsonlite 1.6 2018-12-07 [1] CRAN (R 3.6.0)
knitr 1.24 2019-08-08 [1] CRAN (R 3.6.0)
labeling 0.3 2014-08-23 [1] CRAN (R 3.6.0)
later 0.8.0 2019-02-11 [1] CRAN (R 3.6.0)
lattice 0.20-38 2018-11-04 [1] CRAN (R 3.6.0)
lazyeval 0.2.2 2019-03-15 [1] CRAN (R 3.6.0)
limma * 3.40.6 2019-07-26 [1] Bioconductor
locfit 1.5-9.1 2013-04-20 [1] CRAN (R 3.6.0)
lubridate 1.7.4 2018-04-11 [1] CRAN (R 3.6.0)
magrittr * 1.5 2014-11-22 [1] CRAN (R 3.6.0)
MASS 7.3-51.4 2019-03-31 [1] CRAN (R 3.6.0)
Matrix 1.2-17 2019-03-22 [1] CRAN (R 3.6.0)
matrixStats 0.54.0 2018-07-23 [1] CRAN (R 3.6.0)
memoise 1.1.0 2017-04-21 [1] CRAN (R 3.6.0)
mgcv 1.8-28 2019-03-21 [1] CRAN (R 3.6.0)
mime 0.7 2019-06-11 [1] CRAN (R 3.6.0)
modelr 0.1.5 2019-08-08 [1] CRAN (R 3.6.0)
msigdbr * 7.0.1 2019-09-04 [1] CRAN (R 3.6.0)
munsell 0.5.0 2018-06-12 [1] CRAN (R 3.6.0)
nlme 3.1-141 2019-08-01 [1] CRAN (R 3.6.0)
org.Dr.eg.db * 3.8.2 2019-11-20 [1] Bioconductor
org.Hs.eg.db * 3.8.2 2019-11-20 [1] Bioconductor
pander * 0.6.3 2018-11-06 [1] CRAN (R 3.6.0)
pillar 1.4.2 2019-06-29 [1] CRAN (R 3.6.0)
pkgbuild 1.0.6 2019-10-09 [1] CRAN (R 3.6.0)
pkgconfig 2.0.2 2018-08-16 [1] CRAN (R 3.6.0)
pkgload 1.0.2 2018-10-29 [1] CRAN (R 3.6.0)
plyr * 1.8.4 2016-06-08 [1] CRAN (R 3.6.0)
polyclip 1.10-0 2019-03-14 [1] CRAN (R 3.6.0)
prettyunits 1.0.2 2015-07-13 [1] CRAN (R 3.6.0)
processx 3.4.1 2019-07-18 [1] CRAN (R 3.6.0)
progress 1.2.2 2019-05-16 [1] CRAN (R 3.6.0)
promises 1.0.1 2018-04-13 [1] CRAN (R 3.6.0)
ProtGenerics 1.16.0 2019-05-02 [1] Bioconductor
ps 1.3.0 2018-12-21 [1] CRAN (R 3.6.0)
purrr * 0.3.3 2019-10-18 [1] CRAN (R 3.6.0)
R6 2.4.0 2019-02-14 [1] CRAN (R 3.6.0)
rappdirs 0.3.1 2016-03-28 [1] CRAN (R 3.6.0)
Rcpp * 1.0.2 2019-07-25 [1] CRAN (R 3.6.0)
RCurl 1.95-4.12 2019-03-04 [1] CRAN (R 3.6.0)
readr * 1.3.1 2018-12-21 [1] CRAN (R 3.6.0)
readxl 1.3.1 2019-03-13 [1] CRAN (R 3.6.0)
remotes 2.1.1 2020-02-15 [1] CRAN (R 3.6.0)
RJSONIO 1.3-1.4 2020-01-15 [1] CRAN (R 3.6.0)
rlang 0.4.4 2020-01-28 [1] CRAN (R 3.6.0)
rmarkdown 1.15 2019-08-21 [1] CRAN (R 3.6.0)
rprojroot 1.3-2 2018-01-03 [1] CRAN (R 3.6.0)
Rsamtools 2.0.0 2019-05-02 [1] Bioconductor
RSQLite 2.1.2 2019-07-24 [1] CRAN (R 3.6.0)
rstudioapi 0.10 2019-03-19 [1] CRAN (R 3.6.0)
rtracklayer 1.44.3 2019-08-24 [1] Bioconductor
rvest 0.3.4 2019-05-15 [1] CRAN (R 3.6.0)
rWikiPathways * 1.4.1 2019-07-30 [1] Bioconductor
S4Vectors * 0.22.0 2019-05-02 [1] Bioconductor
scales * 1.0.0 2018-08-09 [1] CRAN (R 3.6.0)
sessioninfo 1.1.1 2018-11-05 [1] CRAN (R 3.6.0)
shiny 1.3.2 2019-04-22 [1] CRAN (R 3.6.0)
stringi 1.4.3 2019-03-12 [1] CRAN (R 3.6.0)
stringr * 1.4.0 2019-02-10 [1] CRAN (R 3.6.0)
SummarizedExperiment 1.14.1 2019-07-31 [1] Bioconductor
testthat 2.3.1 2019-12-01 [1] CRAN (R 3.6.0)
tibble * 2.1.3 2019-06-06 [1] CRAN (R 3.6.0)
tidygraph * 1.1.2 2019-02-18 [1] CRAN (R 3.6.0)
tidyr * 0.8.3 2019-03-01 [1] CRAN (R 3.6.0)
tidyselect 0.2.5 2018-10-11 [1] CRAN (R 3.6.0)
tidyverse * 1.2.1 2017-11-14 [1] CRAN (R 3.6.0)
tweenr 1.0.1 2018-12-14 [1] CRAN (R 3.6.0)
usethis 1.5.1 2019-07-04 [1] CRAN (R 3.6.0)
vctrs 0.2.0 2019-07-05 [1] CRAN (R 3.6.0)
viridis 0.5.1 2018-03-29 [1] CRAN (R 3.6.0)
viridisLite 0.3.0 2018-02-01 [1] CRAN (R 3.6.0)
whisker 0.4 2019-08-28 [1] CRAN (R 3.6.0)
withr 2.1.2 2018-03-15 [1] CRAN (R 3.6.0)
workflowr 1.6.0 2019-12-19 [1] CRAN (R 3.6.0)
xfun 0.9 2019-08-21 [1] CRAN (R 3.6.0)
XML 3.98-1.20 2019-06-06 [1] CRAN (R 3.6.0)
xml2 1.2.2 2019-08-09 [1] CRAN (R 3.6.0)
xtable 1.8-4 2019-04-21 [1] CRAN (R 3.6.0)
XVector 0.24.0 2019-05-02 [1] Bioconductor
yaml 2.2.0 2018-07-25 [1] CRAN (R 3.6.0)
zeallot 0.1.0 2018-01-28 [1] CRAN (R 3.6.0)
zlibbioc 1.30.0 2019-05-02 [1] Bioconductor
[1] /Library/Frameworks/R.framework/Versions/3.6/Resources/library